
Clinical Trial Management
At TedMed Engineering Projects (TMEP) Consulting, we provide comprehensive clinical trial management services that support sponsors, CROs, research sites, and healthcare organizations throughout the entire study lifecycle. Our focus is to ensure studies are conducted efficiently, ethically, and in full compliance with regulatory requirements while maintaining high standards for data quality and patient safety.
Preclinical, Phase I–IV & Post-Market Surveillance Support
Developing a new drug to treat disease is a complex, costly, and time-intensive process that requires strong scientific leadership and disciplined project management. From early discovery through post-market monitoring, development can take more than a decade and requires significant financial investment. Because each phase builds on the success of the previous one, delays, compliance gaps, or poor coordination can lead to major setbacks. This is why having an experienced and trusted consulting partner is essential.
At TedMed Engineering Projects (TMEP) Consulting, our clients rely on us to help guide and strengthen the progression of their clinical trials across the full development lifecycle. Our founder’s many years of industry experience have helped build strong professional alliances with seasoned clinical research experts, creating a collaborative network that brings deep technical knowledge and operational strength to every engagement.
Together with our partners, TMEP provides expertise across key therapeutic areas and innovation-driven fields, including hematology/oncology, chronic disease research, immuno-oncology, cell and gene therapy, biomarker development, and rare disease drug programs. We support research and development teams with structured, compliant, and efficient project management strategies that help move studies forward with confidence.
Our team maintains extensive training in CITI Good Clinical Practice (GCP), as well as Good Manufacturing Practice (GMP) and Good Laboratory Practice (GLP). This foundation allows us to effectively support and manage activities across all phases of drug development, including preclinical planning, Phase I–III clinical trials, Phase IV studies, and post-market surveillance. We also assist with monitoring, auditing, regulatory coordination, data validation, and support for commercial GMP environments.
At every stage of the drug development process, TMEP brings the expertise needed to help design, plan, execute, and control risks within clinical trials. Our goal is to ensure strong regulatory compliance, high-quality data integrity, and efficient study progression from early research through post-market monitoring.
What We Do
Study Start-Up & Planning
• Protocol review and feasibility assessments
• Site identification and readiness evaluation
• Regulatory document preparation and submission support
• Study timelines, budgets, and resource planning
• IRB/ethics committee coordination
Clinical Project Management
• End-to-end oversight of clinical trials from initiation to close-out
• Study timeline tracking and milestone management
• Risk identification and mitigation planning
• Sponsor and stakeholder coordination
• Vendor and CRO oversight
Regulatory Compliance & Documentation
• Ensure compliance with ICH-GCP, FDA, and ISO standards
• Maintain Trial Master File (TMF) and essential documents
• Support regulatory submissions and study updates
• Audit readiness and inspection preparation
Site Management & Support
• Site training and onboarding
• Monitoring study performance and enrollment targets
• Ensuring protocol adherence
• Supporting clinical coordinators and investigators
Quality & Data Integrity Oversight
• Ensure accurate data collection and documentation
• Support query resolution and data review processes
• Implement quality control measures to reduce protocol deviations
• Assist with CAPA development when needed
Equipment & Clinical Operations Support
• Management of clinical trial equipment qualification and calibration
• Coordination of device accountability and tracking
• Support for investigational product/device handling compliance
Patient Safety & Compliance
• Ensure informed consent processes are followed
• Support adverse event reporting and safety documentation
• Maintain confidentiality and ethical research practices
Study Close-Out
• Site close-out coordination
• Final documentation review and archiving
• Lessons learned and process improvement reporting
Our Role
TMEP serves as a bridge between sponsors, research sites, regulatory bodies, and clinical teams. We bring strong expertise in project management, quality systems, regulatory compliance, and clinical operations to ensure trials are completed on time, within budget, and in full compliance.
We specialize in supporting:
• Medical device clinical trials
• Drug and biologics research studies
• Investigator-initiated studies
• Site management and operational readiness
Our goal is to help organizations run structured, compliant, and successful clinical trials that produce reliable results while protecting patient safety.
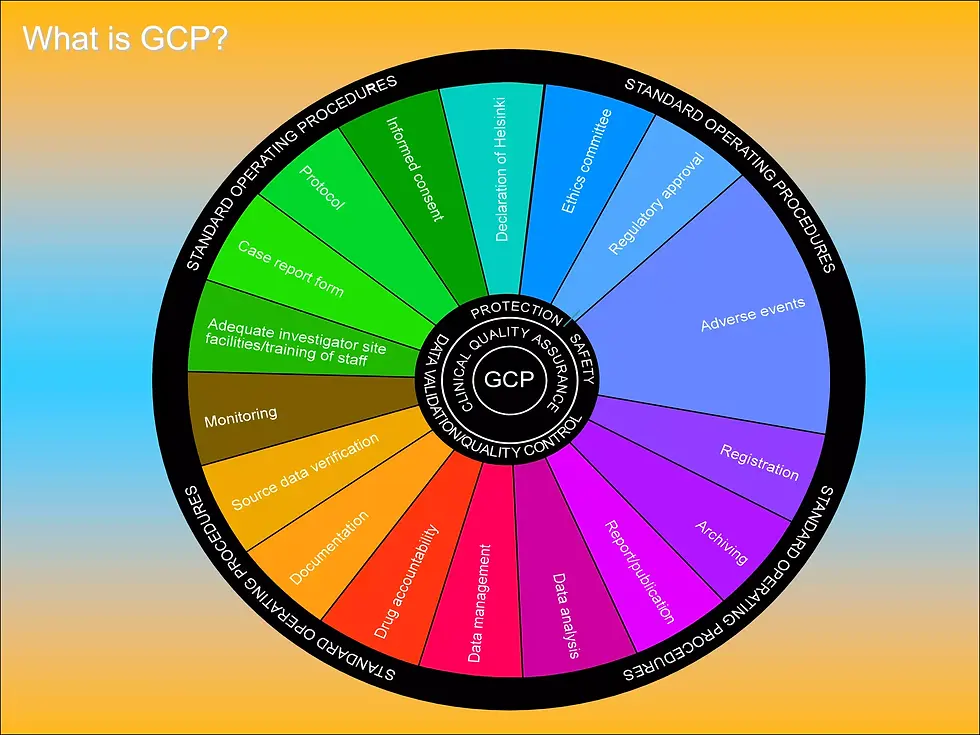
Good Clinical Practice (GCP)
Good Clinical Practice (GCP) is an internationally recognized ethical and scientific quality standard for designing, conducting, recording, and reporting clinical trials involving human participants. It ensures that clinical research is performed with integrity, that participants’ rights and safety are protected, and that the data generated is accurate and credible.
GCP guidelines are based on global regulatory frameworks such as ICH-GCP E6 and are required by regulatory authorities including the FDA and international health agencies. Compliance with GCP is essential for pharmaceutical, biotechnology, and medical device organizations conducting clinical research.
----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
What GCP Means in Practice
GCP focuses on the following core principles:
• Protecting the rights, safety, and well-being of study participants
• Ensuring informed consent is properly obtained and documented
• Maintaining accurate, complete, and verifiable clinical data
• Following approved study protocols and regulatory requirements
• Ensuring qualified personnel manage and conduct studies
• Maintaining proper documentation, monitoring, and audit readiness
These standards apply across the entire clinical trial lifecycle—from planning and site selection to study execution and reporting.
⸻--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------===
What TMEP Consulting Does in Good Clinical Practice (GCP)
___________________________________________________________________________________________________
TedMed Engineering Projects (TMEP) Consulting provides specialized GCP consulting and clinical project management services to support organizations involved in clinical research, including clinical trial sites, sponsors, and medical device or pharmaceutical companies.
Clinical Trial Project Management
TMEP supports end-to-end clinical study coordination by:
• Managing clinical trial timelines, scope, and deliverables
• Coordinating with investigators, sponsors, and CROs
• Supporting study startup activities and regulatory readiness
• Ensuring adherence to protocol and GCP requirements
GCP Compliance Support
TMEP helps organizations establish and maintain strong compliance programs by:
• Training clinical staff on GCP principles and responsibilities
• Ensuring proper informed consent processes
• Supporting protocol implementation and compliance monitoring
• Maintaining regulatory documentation and trial master files (TMF)
Quality Assurance & Audit Readiness
TMEP strengthens research quality systems through:
• Internal GCP audits and inspection readiness preparation
• Gap assessments for clinical sites and study processes
• CAPA development and implementation
• SOP development aligned with ICH-GCP standards
Clinical Site Operations Support
TMEP works directly with clinical sites to ensure operational excellence by:
• Supporting site setup and qualification
• Managing study documentation and records
• Assisting with monitoring visit preparation
• Ensuring accurate and timely data collection
Regulatory & Documentation Management
TMEP ensures that all clinical activities meet regulatory expectations by:
• Maintaining essential documents and regulatory binders
• Supporting IRB submissions and approvals
• Ensuring compliance with FDA and international research standards
• Managing study reports and documentation lifecycle
⸻-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
Value TMEP Brings to Clinical Research
Through your leadership as a Regulatory Consultant, Clinical Project Manager, and Quality & Validation Engineer, TMEP Consulting brings a structured, compliance-driven approach to clinical research. The organization helps:
• Reduce regulatory risks
• Improve study quality and data integrity
• Ensure patient safety and ethical conduct
• Strengthen audit and inspection readiness
• Improve operational efficiency at clinical sites
TMEP acts as both a strategic partner and hands-on execution team, helping research organizations maintain full GCP compliance while successfully managing clinical trials from start to finish.
_____________________________________________________________________________________________________
Learn more about Drug Development Clinical Trial.
Companies cannot market new drugs, medical devices, or biological products, without providing to the U.S. Food and Drug Administration (FDA) proof that the product is safe. Clinical trials are conducted to show safety, and efficacy of the product. Other countries, also have regulatory agencies that require similar proof of new drug safety and efficacy.
Regulatory agencies assess the information and data provided by sponsors and investigators, and then decide to approve or reject the marketing application for the product.
The International Council for Harmonization (ICH) E6 standards, has been implemented in the U.S., Europe, Japan, and other countries, to aid in compliance with regulatory requests of many of governmental regulatory agencies.
ICH E6 R1, and its 2016 addendum R2, relates to conducting clinical trials of drugs, medical device and biologics that abide by Good Clinical Practice (GCP) standards. Like many other to regulations and policies, the ICH E6 guidelines are periodically reviewed.
While many believe that most sponsors are academic and institutional entities within the Biotech / Pharmaceutical industry, that is not always the case. There are also individual investigators, who are sponsors and can also seek FDA approval to study an investigational drug or device. They are known as sponsor-investigators. Sponsor investigators are also required to follow regulatory guidelines just like all investigators and research sponsors.
The Pharmaceutical, Healthcare and Biotechnology industry conduct clinical trials for many different purposes. The number one objective of clinical trials is to show the safety and efficacy of the product, to obtained Food and Drug Administration (FDA) approval. However, there are many clinical trials that are for academic purposes and are not conducted for FDA submission. Nevertheless, those trials are still required to follow FDA And GCP guidelines.
FDA assist researchers on what constitutes as an acceptable clinical trials and appropriate outcomes. However, following FDA regulation is not enough. Hence, many Sponsors and contracted CRO's prefer following ICH GCP guidelines in combination with FDA guidelines to conduct research trials. It is important to note that ICH GCP guidelines are merely a standard in the US and not an enforceable law. Nonetheless, all sponsors require their trials to be conducted in a manner that follows both FDA and ICH GCP standards.
GCP standards are broader than FDA guidelines. ICH, 2016 E6 GCP guidelines, Section 5.0 states that the sponsor should create a system to achieve quality in all phases of the clinical trial. The quality management system should use a risk-based method. This was an essential change for trial monitoring, that extends beyond just routine periodic record auditing. For example, the new ICH E6 integrated addendum (R2), requires sponsors to implement a risk-based process that identifying risks in the study. Sponsors are also required to perform a root case analysis and also CAPA, Corrective Action and Preventative Action when needed.
Section 4.9.0 of ICH E6 also requires the investigator to sustain adequate and accurate source documents and trial proceedings including all related notes on each of the site’s trial participants. Paper, or electronic source data must follow ALCOA-C. This means it needs to be attributable, legible, contemporaneous, original, accurate, and complete. Any changes to source data should be traceable, it shouldn’t obscure the original entry, and must explained if needed.
ICH E6, Section 5.5 and FDA regulations are equally in agreement that the sponsor is accountable for the trial managing, data management, and record keeping. ICH E6 Section 5.5.3 further states that the sponsor must confirm that the electronic trial data handling and/or remote electronic trial data systems follow the sponsor’s written requirements for completeness, accuracy, reliability, and are validated. Written requirement should be a written standard operating procedure also know SOPs.
It is the responsibility of the Sponsor's / CRO's to hire a Clinical Research Professionals who are very experienced in other areas to maintain cost.
Contact us to not only perform monitoring, and auditing your study trials, but to also validate all your regulatory electronic data.